
SMA Awareness Month 1: What is SMA?
What is SMA?
SMA (Spinal Muscular Atrophy) refers to a family of genetic diseases characterized by the degeneration of alpha motor neurons, which facilitate the activity of skeletal muscles, and the resultant underdevelopment and wasting (atrophy) of skeletal muscles. Although the symptoms mainly manifest in muscles related to voluntary action, breathing and swallowing abilities of patients may also be hindered in severe cases [1] [2] [3].
It is important to note that SMA class diseases may be caused by impairments in different genes. Depending on the genomic location of the dysfunctional gene, the disease may be even inherited in autosomal recessive or autosomal dominant manner [2]. When you hear people in the media or elsewhere talking about SMA and the most expensive drug in the world, namely Zolgensma, they are most definitely referring to the class of SMA diseases that are underlied by some dysfunction in the SMN1 gene. Also referred to as “classical proximal SMA”, this class of SMA diseases make up 95% of all SMA cases [4]. Throughout the rest of this text, I will refer to this class of SMA diseases simply as “SMA”, unless explicitly noted.
What causes SMA?
SMA is caused by mutations on or the homozygous deletion of the smn1 gene located at the 5q13 region and the consequent deficiency of SMN (Survival Motor Neuron) protein [5]. Although the whole map of functions of the SMN protein throughout the human body have not been fully elucidated, its ubiquity in all eukaryotic cells and vitality in various metabolic and signalling pathways, such as neurotransmitter release in neurons, in humans underlie the severity of the disease.
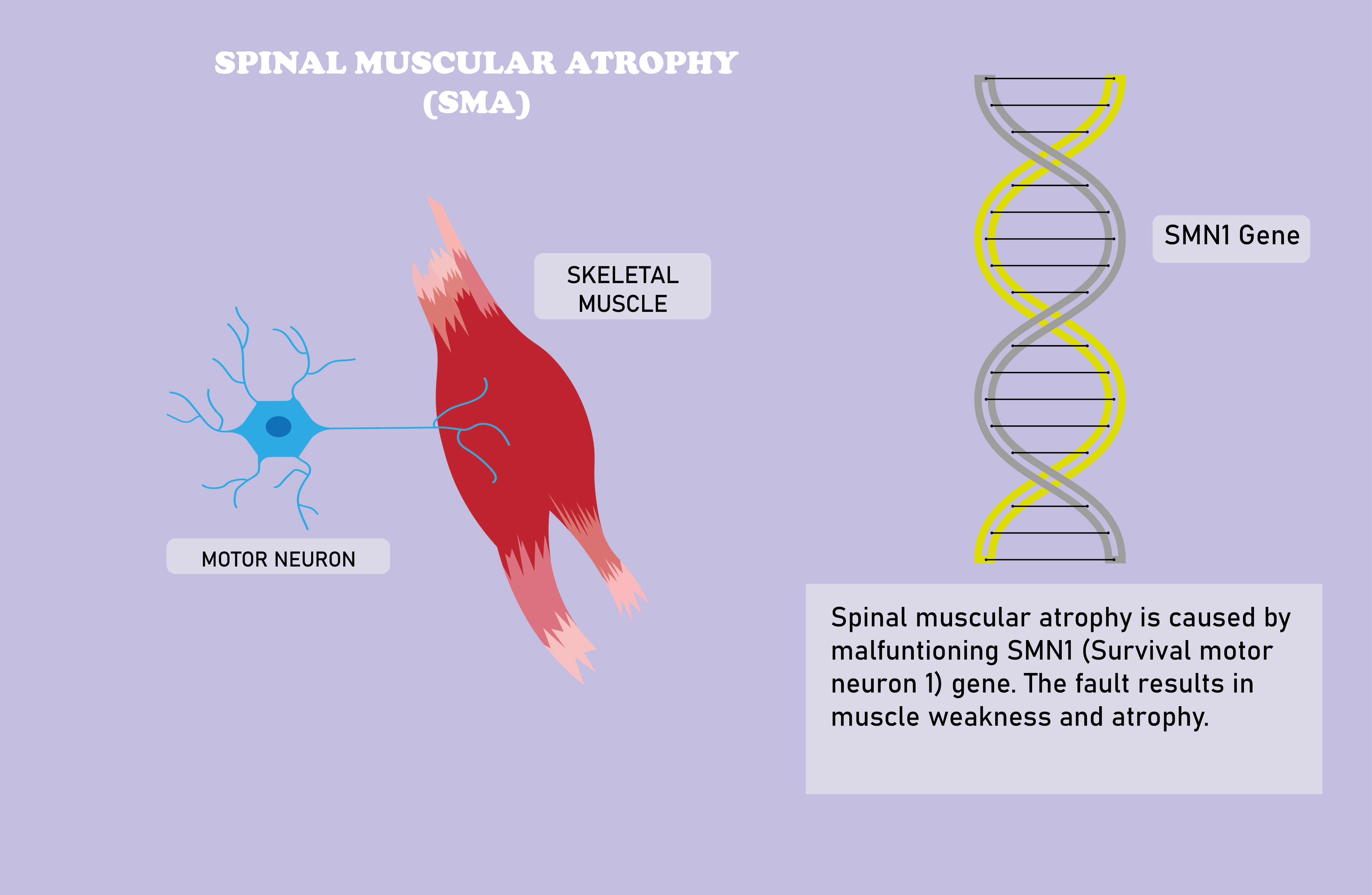
Moreover, it is worth mentioning now that although the SMA family of diseases are defined by damage to alpha motor neurons and muscular atrophy, the prevalence of vital functions of the SMN protein, together with clinical evidence of cases that show symptoms in different systems including but not limited to cardiovascular and reproductive systems, suggests that SMA may be considered as a systemic disease [6].
What are the types of SMA ?
All SMA cases are caused by the inability of the smn1 gene to produce the functional SMN protein, yet the cases can be classified into five distinct phenotypes, from type 0 to type 4. What distinguishes between the different types and the degree of severity of the symptoms is the number of copies of the smn2 gene. which is located in the same chromosome as smn1, but due to a single point difference with smn1 in the nucleotide sequence (the sequence of letters of the genetic code) produces the functional SMN protein with only 10-15% chance.
For non-biologists, I should note that what defines the protein to be produced by a gene is the nucleotide sequence, and that sometimes this sequence also affects how the protein will be modified before being released into the cell environment. In fact, both the smn1 and smn2 genes describe the same protein through their nucleotide sequences, which accounts for the fact that the smn2 gene is able to produce the functional SMN protein. The natural question that arises is why the protein is functional only at 10-15% of the time, and this is where the single nucleotide difference comes into play. Because, apparently at 85-90% of the time, it triggers a change in the structure of the SMN protein after it is initially produced from the template of the nucleotide sequence, which renders the protein prone to being dysfunctional and short-lived. Thus, one can think of the smn2 gene as a rather clumsy duplicate of smn1, failing to live up to the standards of its hard working colleague.
Back to SMA. As I noted earlier, the number of copies of the smn2 gene in the patients’ genome is crucial for it defines the types of the disease. As you may have guessed, simply put, the more smn2 gene a patient has, the lesser the symptoms will be.
Below is a brief list of clinical characterizations of types of SMA. [4]
Type 0: Also called congenital SMA, this is a very rare phenotype. The onset period is prenatal, and patients do not live past the first months of life, with very severe symptoms of proximal atrophy and respiratory or circulatory complications.

Type 1: Also known as Werdnig-Hoffmann Disease, a homage to the clinicians that first systematically observed this type of SMA in late 1890s, or severe SMA, Type I is defined by an early onset in the first six months of life, and symptoms include severe underdevelopment of skeletal muscles and difficulty in respiration and swallowing. Life expectancy is around two years if the patient is not provided ventilation support.
Type 2: Also called Dubowitz disease or intermediate SMA, Type 2 SMA is characterized by onset between 6-18 months and less severe muscular symptoms. Patients may sit, but are restricted in skeletal and muscular growth. Most live until 25, and the most frequent cause of mortality is respiratory complications related to restrictive lung disease induced by scoliosis and muscular atrophy.
Type 3: Also termed Kugelberg-Welander Syndrome or mild SMA, Type 3 disease is characterized by Type 2-like symptoms of progressive proximal atrophy in skeletal muscles, yet the patients are usually able to walk. Life expectancy is not significantly affected.
Type 4: Also called adult SMA, Type 4 is the mildest phenotype. Symptoms appear during adulthood and patients develop only mild progressive proximal weakness. Life expectancy is not significantly affected. [4]
How is SMA diagnosed?
In case of any suspicion raised by the characteristic symptoms of the disease, genetic testing through simple blood tests are able to diagnose smn1 related SMA cases with very high accuracy. Since the genes that underlie the disease are definitively characterized, and genetic testing methods such as polymerase chain reaction are well established, diagnosis through genetic testing is highly reliable, with accuracy rates 95% or higher.[1] [4]
Such reliable methods prove to be quite critical also in the prevention of the disease. Since dysfunctions of the smn1 gene are inherited in an autosomal recessive manner, which implies that the disease may only occur if both parents are carriers for the disease, informing healthy individuals about whether they are carriers or not through simple and accurate genetic testing methods proves to be a very powerful tool in preventing the occurrence of the disease.
Other diagnostic methods include electromyography, nerve conduction velocity studies, and muscular biopsy, which prove valuable in validating the diagnosis of the disease in cases where the underlying genetic cause is not related to any of the well-studied genotypes or genetic testing fails to validate the diagnosis. [1]
Topics we did not cover in this post, especially that of treatments of SMA, will be elaborated on in our next post. Stay tuned.
REFERENCES
- Office of Communications and Public Liaison. (2019, May). Spinal Muscular Atrophy Fact Sheet, National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Spinal-Muscular-Atrophy-Fact-Sheet
- Spinal Muscular Atrophy, National Organisation for Rare Diseases. https://rarediseases.info.nih.gov/diseases/7674/spinal-muscular-atrophy#ref_2299
- Keinath, M. C., Prior, D. E., & Prior, T. W. (2021). Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. The application of clinical genetics, 14, 11–25. https://doi.org/10.2147/TACG.S239603
- Burr, P., & Reddivari, A. (2021). Spinal Muscle Atrophy. In StatPearls. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK560687/
- Kolb, S. J., & Kissel, J. T. (2011). Spinal muscular atrophy: a timely review. Archives of neurology, 68(8), 979–984. https://doi.org/10.1001/archneurol.2011.74
- Lipnick, S. L., Agniel, D. M., Aggarwal, R., Makhortova, N. R., Finlayson, S. G., Brocato, A., Palmer, N., Darras, B. T., Kohane, I., & Rubin, L. L. (2019). Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PloS one, 14(3), e0213680. https://doi.org/10.1371/journal.pone.0213680
- Sugarman, E., Nagan, N., Zhu, H. et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Eur J Hum Genet 20, 27–32 (2012). https://doi.org/10.1038/ejhg.2011.134